

FDA Expedited Drug Development Programs
The Food and Drug Administration (FDA) follows an established and lengthy approval process that ensures patients have access to therapeutic agents proven to be safe and effective. The process relies upon a structured framework that includes an analysis of the target condition and available treatments, an assessment of benefits and risks from clinical trials, and strategies for managing these risks.
When a therapy treats life-threatening conditions, addresses an unmet need, or provides therapeutic benefit over existing therapies, the Food and Drug Administration (FDA) drug approval process can be expedited. To make drugs accessible as soon as possible to patients, the FDA offers the following designations:
- FDA Accelerated Approval – Allows drugs for serious conditions that fill an unmet need to be approved based on a surrogate endpoint
- Fast Track – Facilitates the development and expedites the review of drugs that treat a serious condition or meet an unmet need
- Breakthrough Therapy – Expedites the development and review of drugs that demonstrate substantial improvement over available therapies
The ability to shorten the drug development and approval process has huge implications for drug manufacturers and patients suffering from serious conditions. Having insight into which therapies have received one of these designations and which therapeutic areas have been awarded the most designations can be extremely helpful to manufacturers when developing clinical trials, business development, and partnership strategies.
In the last three years alone, there have been over 350 FDA Accelerated Approval, Breakthrough Therapy, and Fast Track designations awarded across over 50 disease areas; for a full list, download the PDF at the bottom of the page.
Fast Track
FDA Fast Track designation is designed to get new drugs to patients who have serious conditions or that treat an unmet medical need. It covers a broad range of conditions and is based on whether the drug will have an impact on survival, day-to-day functioning, or the likelihood that the condition will progress to a more serious one.
The Fast Track designation has been critical for patients suffering from Alzheimer’s, cancer, HIV/AIDS, epilepsy, depression, diabetes, and heart failure. It has accelerated access to therapies that have helped to slow the progression of these conditions.
When there are already available therapies in the market that treat a serious condition, drugs seeking a Fast Track designation must meet several criteria to demonstrate an advantage over these. The criteria include:
- Demonstrating superior efficacy
- Avoiding serious side effects
- Improving diagnosis when the patient is diagnosed early
- Decreasing significant toxicity that may lead to discontinuation
- Addressing an emerging or anticipated health need
Most importantly, the Fast Track designation results in more frequent and earlier communication between the FDA and the manufacturer throughout the drug development and review process. This includes more frequent meetings with the FDA to discuss the development plan and to ensure appropriate data for approval is being collected, more frequent written communication with the FDA regarding the design of trials and use of biomarkers, eligibility for Accelerated Approval and Priority Review designations if relevant criteria are met, and Rolling Reviews, where sections of a New Drug Application (NDA) or Biologic License Application (BLA) can be reviewed by the FDA once completed instead of waiting until the entire application is officially submitted.
A request for a Fast Track designation can be made at any time but must be done by the drug manufacturer. Manufacturers can expect a response from the FDA within 60 days after submittal. Once a drug receives this designation, the manufacturer is highly encouraged to maintain early and frequent communication with the FDA to make sure issues are addressed immediately, which can assist with earlier drug approval and distribution to patients.
Breakthrough Therapy
To receive a Breakthrough Therapy designation, a drug must demonstrate substantial improvement over an existing therapy on a clinically significant endpoint based on preliminary clinical evidence. The treatment must be for a serious condition, and the clinically significant endpoint must impact irreversible morbidity, mortality, or symptoms that signify serious consequences of the disease.
Determining improvement over another therapy depends on the magnitude of the effect and the importance of the observed clinical outcome. Ultimately, there should be a substantial improvement in preliminary clinical evidence on clinically significant endpoints such as an established surrogate endpoint, an intermediate clinical endpoint that predicts a clinical benefit, a pharmacodynamic biomarker that strongly suggests a clinically meaningful effect on the disease, or a significantly improved safety profile compared to the available therapy.
Drugs that receive this designation are eligible for the same accelerated communication and review options that Fast Track designations receive, as well as involvement by FDA senior management and intensive guidance on how to create an efficient drug development program as early as phase 1. A manufacturer can request a Breakthrough Therapy designation no later than the end of phase 2, but if a drug development program meets the Breakthrough Therapy criteria and the remaining development program can benefit from the designation, the FDA may also suggest that the manufacturer submit a request. Breakthrough Therapy designation requests receive responses from the FDA within 60 days of receipt.
Accelerated Approval
Since 1992, manufacturers have been able to pursue Accelerated Approval regulations, which allow for the approval of a drug before it completes all phases of its drug development program if it demonstrates an effect on:
- A surrogate endpoint* or marker that is reasonably likely to predict clinical benefit
- An intermediate endpoint† that occurs earlier and may not be as robust as the study endpoint
* Surrogate endpoint is a laboratory measurement, a radiographic image, or a physical or other sign that is thought to predict a clinical benefit
† Intermediate endpoint is a measure of a therapeutic effect that is considered reasonably likely to predict the clinical benefit of the drug, such as an effect on irreversible morbidity and mortality
Accelerated Approval follows the same framework as the traditional drug approval process but saves valuable time by basing approval on earlier trial results. An example of this is Gleevec, an oral treatment for patients with chronic myeloid leukemia (CML) that was approved under the FDA’s Orphan Drug program. Gleevec received Accelerated Approval after submitting the results of three large phase 2 trials.
Receiving approval based on phase 2 trials or early results from phase 3 trials can be extremely beneficial for patients suffering from conditions for which it can take years to measure a drug’s intended clinical benefit. Studies that demonstrate a drug’s effect on one of the endpoints listed above must be “adequate and well-controlled” as required by the Food, Drug, and Cosmetic (FD&C) Act. Once a result that is reasonably likely to predict a real clinical benefit has been achieved, a drug can receive Accelerated Approval.
An important consideration regarding this approach is that manufacturers must conduct post-marketing or phase 4 confirmatory trials to verify the drug’s benefit after it enters the market. When these trials confirm clinical benefit, the FDA will grant full approval. If these trials fail or do not demonstrate that the benefits outweigh the risks of the drug, the FDA will withdraw approval or request that the indication in the drug’s label be changed.
Similar Designations in the EU
The European Medicines Agency (EMA) also has designations that accelerate a patient’s access to drugs that address unmet medical needs. Accelerated Assessment and Conditional Marketing Authorisation are intended for innovative medicines that target a disease that has no available treatment, provide a major therapeutic advantage over existing therapies, or are intended for use in emergency situations to avert a public health crisis.
The guidelines for Accelerated Assessment require applicants to provide detailed guidance on how the drug’s approval is one of major public health interest. The application must explain how the drug addresses an unmet medical need in prevention, diagnosis, or treatment. The evaluation time is shortened to 150 days from 210 days for regular submissions, and drug manufacturers are encouraged to initiate dialogue with the agency early to help plan for Accelerated Assessment.
Conditional Marketing Authorisation allows for the approval of a drug based on less complete clinical data than what is normally required if the drug addresses an unmet medical need, targets a seriously debilitating or life-threatening disease, or is intended for use in emergency situations due to a public health threat. The data used for Conditional Marketing Authorisation must demonstrate that the benefits outweigh the risks of the drug and that the full data can be submitted within a specified timeframe agreed to by the manufacturer and the Committee for Medicinal Products for Human Use (CHMP).
The Impact on Manufacturers and the Market
The FDA has been under pressure over Accelerated Approvals, as numerous drugs cleared via the program have not been confirmed to benefit patients in follow-up testing.To address this, Congress has proposed bills mandating that drug manufacturers must begin confirmatory trials before expediting the drug approval process.
These proposals also grant the FDA the power to specify the design of the confirmatory trials and require manufacturers to report their progress toward completion every 180 days.8 This requirement is meant to address what is known as “dangling” Accelerated Approvals, or drugs that remain on the market with this designation for years without clinical or real-world evidence that they work.
These shifts greatly affect the costs and timing associated with confirmatory testing but are intended to remove drugs that are not showing proof of efficacy in their patient populations. They also point to an evolving regulatory and legislative environment.
For patients and manufacturers, there are several valuable benefits to applying for these designations. The COVID pandemic that began in 2020 is a prime example of how expediting a vaccine or drug’s access to the market can greatly benefit a population. The introduction of HIV/AIDS therapies in the 1990s is also proof that getting therapies to patients who are suffering from terminal illnesses is critical to extending their lives and improving their quality of life. A final reason to continue to pursue these FDA designations is that most oncology drugs, which make up 66% of all FDA Accelerated Approvals since 2000, are eventually fully approved after their confirmatory trials are complete. Only a minority of Accelerated Approvals are withdrawn because the treatment did not demonstrate a benefit in confirmatory trials.
Realizing there is still a strong need to request and attain these FDA designations, manufacturers must have a comprehensive view of the marketplace to stay competitive. Monitoring regulatory milestones of other drug companies carries great benefits as long as it is comprehensive, credible, and tailored to the objectives of the company collecting the data.
The Challenge to Stay Ahead
To remain competitive and make sound strategic business decisions regarding if and when to request one of these FDA designations, companies need to be able to measure how their drugs and their competitors’ drugs are performing in the clinical trial setting. This can be done through an analysis of national and global clinical trial data.
Manufacturers must begin by identifying which companies are being awarded these FDA designations. This will supply a big-picture view of their pipelines and strategies when it comes to expedited approvals. It is also pertinent to identify which therapeutic areas are most active when it comes to these designations. This information can help identify opportunities regarding conditions with unmet needs that are not being addressed or conditions where there is an abundance of compounds already competing for future market share.
A deeper analysis can also help manufacturers determine if existing approved drugs are exploring additional indications, as well as identify which therapies are seeking FDA designations for these line extensions. Through data interpretation, it is possible to determine the launch timing and commercialization strategy of the companies who are seeking any of the three FDA designations.
It is also critical for manufacturers to identify what factors impact the probability that a product will successfully obtain one of these designations. Data analysis can determine which variables did or did not help achieve an FDA designation, which can be very helpful when submitting a request for Fast Track, Breakthrough Therapy, or Accelerated Approval.
Measuring the Impact
OZMOSI’s proprietary pharmaceutical intelligence platform can search the available clinical trial data and efficiently conduct these analyses. Our technologies access, refine, and index global clinical trial data using the latest machine-learning techniques. And by incorporating BEAM, a radically new competitive intelligence tool, we can provide you with the insights you need through intuitive visuals, dynamic views, and custom reports derived from our reporting database of over 600,000 clinical trials across six different clinical trial registries.
We take that data even further with PRYZM, which uses advanced analytics to deliver insights on preclinical, clinical, and approved pharmaceuticals in use worldwide. We pull from approval registries from over 80 countries, more than 70,000 company reports, more than 100,000 PubMed articles, and multiple FDA data sources that identify filing designations for Accelerated Approval, Breakthrough Therapy, Fast Track, and Orphan Drugs.
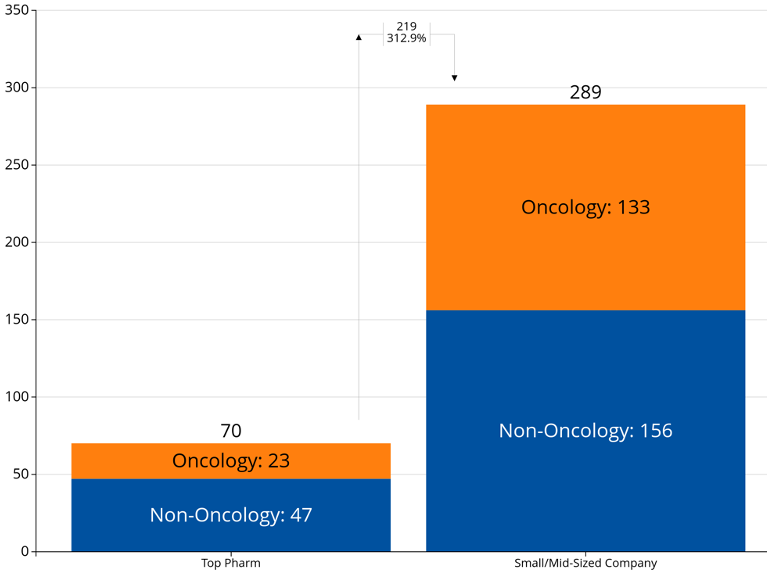
For this project, we leveraged PRYZM to analyze all drugs that received an Accelerated Approval, Breakthrough Therapy, or Fast Track designation from the FDA between 2020 and 2022 (collectively referred to as “FDA designations”). We also used BEAM to analyze the clinical trial implications for drugs that received these designations compared to those without a designation. Our analysis identified that the FDA is disproportionately awarding these designations based on the therapeutic area targeted and the modality of the drug. While many of the top pharmaceutical companies have received multiple FDA designations, we also found that most designations have been awarded to smaller to mid-sized companies, with almost 300 companies appearing on the list with at least one FDA designation (see full PDF list at the bottom of the page).
Among our findings:
- Oncology drugs accounted for nearly half of all FDA designations, with 34% of designations going to drugs targeting solid tumor indications and 10% to drugs targeting hematological tumors. None of the nearly 50 other disease areas accounted for more than 7% of FDA designations.
- Small molecules received only half of FDA designations, with a significant proportion (23%) going to newer modalities such as cell therapies, gene therapies, and antibody drug conjugates (ADCs).
- Only 19% of FDA designations were awarded to large pharmaceutical manufacturers, with smaller manufacturers and biotech companies accounting for the other 81%. Large-cap manufacturers received a higher percentage of Breakthrough Therapy designations (43%) and Accelerated Approval designations (40%).
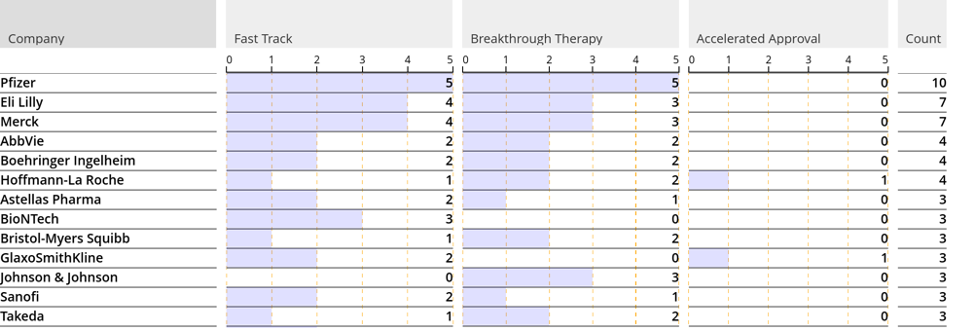
Pfizer, Lilly, and Merck were awarded the most FDA designations, followed by AbbVie, Boehringer Ingelheim, and Roche. There were only 13 companies that received 3 or more FDA designations, and these companies accounted for just 16% of all designations received. Eli Lilly delivering two designations per year for the last three years across multiple disease areas, the most recent one being tirzepatide in obesity. Novartis was the only Top 10 pharmaceutical company to receive only one FDA designation for their CAR-T product tisagenlecleucel.
OZMOSI platforms are fully integrated, so we were able to simply translate our PRYZM product-level analysis into BEAM to analyze any clinical trials that were subsequently initiated after a drug was awarded an FDA designation between 2020 and 2022. We learned that clinical trials in all phases are impacted by an FDA designation, but the largest benefit is observed in phase 2 trials. Our findings included:
- Phase 2 trials accounted for 51% of trials initiated for drugs that had previously received an FDA designation, with the remaining trials evenly split between phase 1 and phase 3
- The estimated probability of success (POS) for these phase 2 trials was 42% across all FDA designations received and even higher (48%) when the designation was Accelerated Approval or Breakthrough Therapy. Estimated phase 1 POS was also higher when the drug had received an Accelerated Approval or Breakthrough Therapy designation.
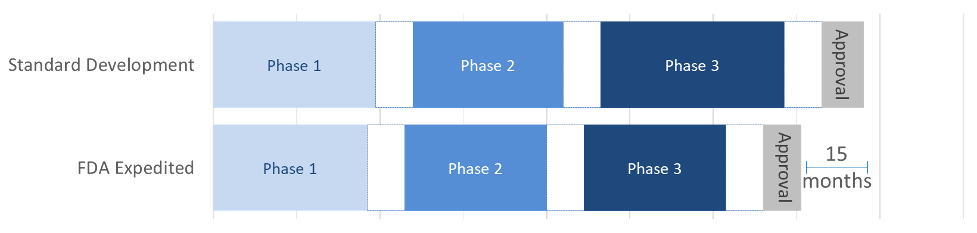
Finally, we investigated whether FDA designations reduce the clinical trial length and time to market. We found that FDA designations accelerated a drug’s first approval and that phase 1 and phase 2 trials were shorter when the study drug had previously received an FDA designation. Our takeaway:
- The median time to first approval for drugs with an FDA designation was 15 months faster than the industry benchmark time to approval. Time to first approval was accelerated even further (21 months) for drugs that had received a Breakthrough Therapy designation.
These results highlight the value of seeking an FDA designation early in the drug development process. The acceleration in time to market for drugs with an FDA designation appears to be driven by shorter phase 1 and 2 trials, together with the possibility for expedited approval associated with an Accelerated Approval or Breakthrough Therapy designation. An FDA designation also reduces early-phase development risk, most conspicuously in phase 2.
Our analysis demonstrates that innovator companies should build clinical development strategies with the intent of applying for an FDA designation as soon as possible to take advantage of the benefit to early phase trial length and development risk. Large pharmaceutical manufacturers can leverage our findings to convince potential licensing and acquisition targets of the value of an early tie-up, particularly when the acquirer’s resources can accelerate an FDA designation application. For biotech companies, our findings show that smaller companies are successfully applying for FDA designations, but they also suggest that having a large-cap partner may be valuable when seeking a Breakthrough Therapy or Accelerated Approval designation.
OZMOSI’s Data Intelligence Platforms Drive Drug Development Strategies
OZMOSI has the data and expertise to support your clinical and commercial strategy development. We apply state-of-the-art techniques to collect, refine, and index data on clinical stage assets from dozens of trusted sources worldwide. Our resulting intelligence platform supports in-depth analyses that are the basis of our clients’ drug development and commercialization strategies.
Through our BEAM and PRYZM platforms, we help biotech and pharmaceutical companies identify development programs and acquisition targets with the potential for pursuing FDA Accelerated Approval, Fast Track, or Breakthrough Therapy designations. Please contact us for more information if you are interested in how OZMOSI’s data intelligence platforms can support your clinical strategy, acquisition strategy or commercial strategy development.

Complete the form below to download a full list of all expedited designations awarded by the FDA by date, company, drug name, designation type, and disease area.
White Paper Download Form
"*" indicates required fields